Quantum Espressoでスーパーセル法のやり方 [Supercell]
1. 概要
QuantumEspresso(以下、QE)は凄い。単位格子でいいなら、市販のPCと非専門家でも第一原理計算ができる。しかし、スーパーセル法に手を出すと話が変わる。少なくとも実行するPCのRAMは16GB以上、計算対象の結晶構造を理解する必要がある。さらに、インターネット上にあるQEのチュートリアルではスーパーセル法についてあまり触れられていない。
本記事では、QEのスーパーセル法の実行方法について説明する。大まかな流れとしては、Siの結晶構造を示すCIFファイルをVESTAを用いて2x2x2に拡張し、pwscfを実行する。
2. 使用ソフトウェアとインストール方法が書いてあるサイト等
|
|


- VESTA
http://www2.yukawa.kyoto-u.ac.jp/~koudai.sugimoto/dokuwiki/doku.php?id=vesta
- cif2cell
http://www2.yukawa.kyoto-u.ac.jp/~koudai.sugimoto/dokuwiki/doku.php?id=quantumespresso:cif2cell
- Xcrysden
http://www2.yukawa.kyoto-u.ac.jp/~koudai.sugimoto/dokuwiki/doku.php?id=xcrysden
3. 大まかな流れ
- cifファイルをVESTAで開き、2x2x2のスーパーセルに展開する。
- VESTAから結晶構造ファイルをxtl形式で出力する。
- 出力したxtlファイルを再度VESTAで開く。
- 今度はcif形式でVESTAから出力する
- cif2cellでpwscf用のインプットファイルに変換する。
- xcrysdenでインプットファイルを開き、構造を確認する。
- インプットファイルの形式を整え計算実行。
3.1 cifファイルをVESTAで開き、2x2x2のスーパーセルに展開する。 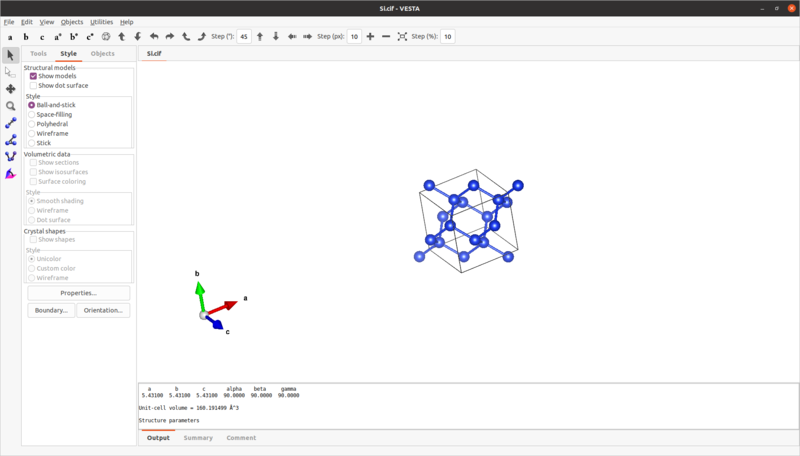
VESTAでSiのcifファイルを開く。VESTAウィンドウ左上の
File -> Open
を押すとファイルブラウザが出てくるので、ここでファイルを選択し開く。
次にVESTAウィンドウ左上の
Edit -> Edit Data -> Unit cell
を押すと、Edit Dataウィンドウが出現する。この状態で、Unit cellタブ内の[Transform...]ボタンをクリックする。

Unit Cell Transformationウィンドウ内のRotation matrix Pの数値を変更することで、単位格子を繰り返し拡張することができる。初期状態では対角成分が1、それ以外が0となっている。対角成分の1行目がx軸方向、2行目がy軸、3行目がz軸方向への拡張となる。(結晶軸としてはx軸=a軸、y軸=b軸、z軸=c軸とする)

今回は2x2x2なので、対角成分にすべて2を入れ、OKボタンを押す。この時に2回の警告が出てくるが、すべてYESで問題ない。


Unit Cell Transformationウィンドウが閉じるのでEdit DataウィンドウのOKを押し、これも閉じる。2x2x2に拡張されたSiの構造が表示されている。

3.2 VESTAから結晶構造ファイルをxtl形式で出力する。
VESTAウィンドウ左上の
File -> Export Data
を押し、出力ファイル形式をxtlに選択する。今回は「Si_2x2x2.xtl」として出力した。

3.3 出力したxtlファイルを再度VESTAで開く。
出力した「Si_2x2x2.xtl」を開く。
3.4 今度はcif形式でVESTAから出力する
今度はcif形式で出力する。このファイルは「Si_2x2x2.cif」とした。
3.5 cif2cellでpwscf用のインプットファイルに変換する。
端末を開き、3.4で出力した「Si_2x2x2.cif」ファイルのディレクトリへ入り、
$ cif2cell -f Si_2x2x2.cif -p pwscf -o Si_2x2x2.in と入力する。同ディレクトリに「Si_2x2x2.in」が出力されていることをファイルブラウザなどで確認する。

ー<なぜこの操作が必要なのか>ーーーーーーーーーーーーーーーーーーーーーーーー
様々なcifファイルはスペースグループを用いて結晶構造を表現している。しかし、cif2cellは単純立方(スペースグループ: P-1)による結晶構造の表現でないと、変換後に出力されるファイルの結晶構造はおかしくなってしまう。
おそらく、VESTAに単純立方への変換機能があるはずなのだが、本記事著者は見つけられなかった。
ーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーーー
3.6 XCrysDenでインプットファイルを開き、構造を確認する。
XCrysDenを起動する。ウィンドウ左上の
Open Structure ... -> Open PWscf input File
を押すとファイルブラウザが出てくるので、3.5で出力した「Si_2x2x2.in」を選択し、Openを押す。
2つほどウィンドウが出てくるが、インプットファイルのテキスト表示のウィンドウは[Close]ボタン、もう一つはチェックボックスはそのままで[OK]ボタンを押す。


XCrysDenがインプットファイルを読み込み、描画された結晶構造が2x2x2のスーパーセルになっているか確認する。確認できたらXCrysDenは閉じる。

3.7 インプットファイルの形式を整えscf計算実行。
「Si_2x2x2.in」をコピーし、「Si_2x2x2.scf.in」とする。テキストエディタを開き「Si_2x2x2.scf.in」を編集する。

ここが意外と難問だ。まず、QEのインプットファイルの記述項目の順は
&CONTROL -> / -> &SYSTEM -> / -> &ELECTRONS -> / -> &CELL_PARAMETERS
-> ATOMIC_SPECIES -> ATOMIC_POSITIONS -> K_POINTS
となる。さらに、末尾のカンマ( , )を付ける場合はすべての行に付け、付けない場合は全部消す必要がある。加えて、ダブルクォーテーション( " )かシングルクォーテーション( ' )のどちらかで統一する。混在すると計算実行時にエラーが生じる。
前回成功したインプットファイルのコピー&ペースト であっても、上記の記述方法から逸脱するとエラーが生じる。著者が発生させた多くのエラーがこれであったため、賢い読者は、まずファイルの記述を確認することをお勧めする。
ファイルの編集後は通常通りに計算を実行する。
 (編集後の例)
(編集後の例)
4. 終わりに
スーパーセル法を用いることでドーピングの影響も評価できる。元素置換型で良いのなら、インプットファイルのATOMIC_POSITIONSでドーピングしたい元素を、元々の原子と入れ替えるだけでよい。その後、ドーピング元素のPSEUDOファイルを参照する必要がある。だが著者が量子力学とかバンド理論とかバンドエンジニアリングとかに詳しくないので、本記事では紹介しない。
本記事執筆中、著者はD1で退学することになった。原因は全実験機器の修理を巡った指導教員との大喧嘩で、最終的に著者の実験費用申請の許可が下りなくなり、自己負担となってしまったためだ。なのでQEの記事を書くことはおそらく無いだろう。乙!